AAV Capsid Structure:
The AAV capsid is made up of three structural proteins: VP1, VP2, and VP3, which self-assemble into a capsid. These proteins have distinct regions and domains that play important roles in virus-cell interactions, receptor binding, and intracellular trafficking. AAV capsids have variations in their surface structures that affect their interactions with host cells and the immune system.
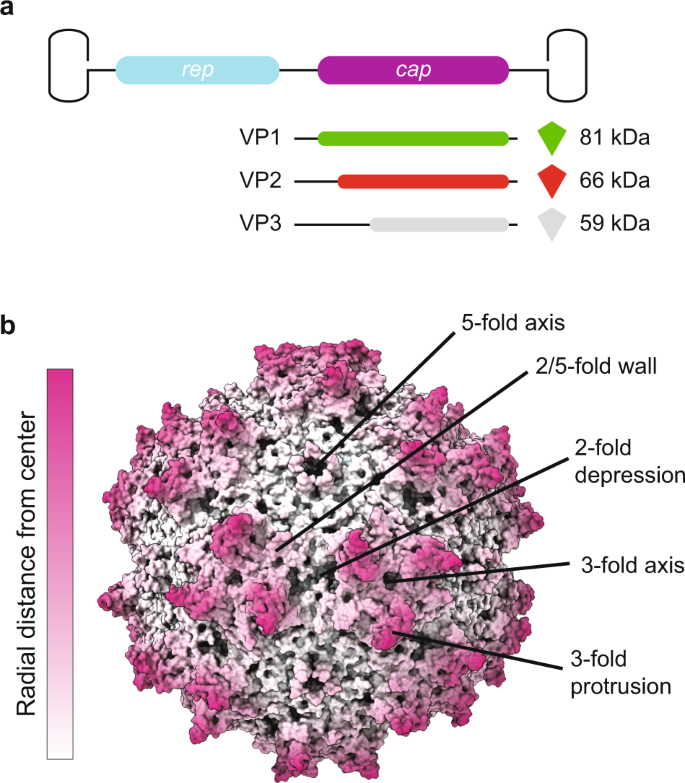
AAV as a Gene Therapy Vector:
rAAV has been used as a gene delivery vector since the early 1980s. It has several advantages, including its simplicity, low immunogenicity, and the ability to transduce a variety of cell types. Different AAV serotypes have tissue-specific preferences, making them suitable for various therapeutic applications.
Rational Design Strategies for AAV Capsid Engineering:
- Scientists use rational design strategies to modify AAV capsids for specific purposes. Three primary approaches are discussed:
- Genetic Mutation: Specific amino acid residues in AAV capsids are mutated to enhance transduction efficiency, specificity, or immune evasion. These mutations can also be used to create capsid chimeras by transferring domains from one serotype to another.
- Insertion of Nonviral Parts: Functional domains or motifs from nonviral sources, like targeting ligands or protease cleavage sites, can be inserted into AAV capsids to impart new functions or responses to stimuli.
- Chemical Biology Approaches: Chemical modifications are used to introduce specific molecules or tags to the capsid, allowing for precise control over capsid properties and interactions.
These rational design strategies aim to improve AAV vectors for gene therapy by enhancing their transduction efficiency, targeting specificity, and other functionalities
Fo reference go to,
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6516759/